Immunofluorescence Analysis as a Diagnostic Tool in a Spanish Cohort of Patients with Suspected Primary Ciliary Dyskinesia

 , , , , , , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Experimental Section
2.1. Patients
2.2. PCD Diagnostic Evaluation
2.3. Immunofluorescence Technique and Analysis
2.4. Data Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lucas, J.S.; Burgess, A.; Mitchison, H.M.; Moya, E.; Williamson, M.; Hogg, C. Diagnosis and Management of Primary Ciliary Dyskinesia. Arch. Dis. Child. 2014, 99, 850–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reula, A.; Lucas, J.S.; Moreno-Galdó, A.; Romero, T.; Milara, X.; Carda, C.; Mata-Roig, M.; Escribano, A.; Dasi, F.; Armengot-Carceller, M. New Insights in Primary Ciliary Dyskinesia. Expert Opin. Orphan Drugs 2017, 5, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile Ciliopathies. Nat. Rev. Dis. Prim. 2020, 6, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Davis, S.D.; Ferkol, T.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Wolf, W.; et al. Laterality Defects Other Than Situs Inversus Totalis in Primary Ciliary Dyskinesia. Chest 2014, 146, 1176–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemark, A.; Frost, E.; Dixon, M.; Ollosson, S.; Kilpin, K.; Patel, M.; Scully, J.; Rogers, A.V.; Mitchison, H.M.; Bush, A.; et al. Accuracy of Immunofluorescence in the Diagnosis of Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2017, 196, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.S.; Barbato, A.; Collins, S.A.; Goutaki, M.; Behan, L.; Caudri, D.; Dell, S.; Eber, E.; Escudier, E.; Hirst, R.A.; et al. European Respiratory Society Guidelines for the Diagnosis of Primary Ciliary Dyskinesia. Eur. Respir. J. 2017, 49, 1601090. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Davis, S.D.; Polineni, D.; Manion, M.; Rosenfeld, M.; Dell, S.D.; Chilvers, M.; Ferkol, T.W.; Zariwala, M.A.; Sagel, S.D.; et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 197, e24–e39. [Google Scholar] [CrossRef]
- Ibañez-Tallon, I.; Heintz, N.; Omran, H. To Beat or Not to Beat: Roles of Cilia in Development and Disease. Hum. Mol. Genet. 2003, 12, R27–R35. [Google Scholar] [CrossRef] [Green Version]
- Olm, M.A.K.; Caldini, E.G.; Mauad, T. Diagnosis of Primary Ciliary Dyskinesia. J. Bras. Pneumol. 2015, 41, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Omran, H.; Loges, N.T. Immunofluorescence Staining of Ciliated Respiratory Epithelial Cells. Methods Cell Biol. 2009, 91, 123–133. [Google Scholar] [CrossRef]
- Liu, Z.; Nguyen, Q.P.H.; Guan, Q.; Albulescu, A.; Erdman, L.; Mahdaviyeh, Y.; Kang, J.; Ouyang, H.; Hegele, R.G.; Moraes, T.J.; et al. A Quantitative Super-Resolution Imaging Toolbox for Diagnosis of Motile Ciliopathies. Sci. Transl. Med. 2020, 12, eaay0071. [Google Scholar] [CrossRef] [Green Version]
- Behan, L.; Dimitrov, B.D.; Kuehni, C.E.; Hogg, C.; Carroll, M.; Evans, H.J.; Goutaki, M.; Harris, A.; Packham, S.; Walker, W.T.; et al. PICADAR: A Diagnostic Predictive Tool for Primary Ciliary Dyskinesia. Eur. Respir. J. 2016, 47, 1103–1112. [Google Scholar] [CrossRef] [Green Version]
- Horváth, I.; Barnes, P.J.; Loukides, S.; Sterk, P.J.; Högman, M.; Olin, A.-C.; Amann, A.; Antus, B.; Baraldi, E.; Bikov, A.; et al. A European Respiratory Society Technical Standard: Exhaled Biomarkers in Lung Disease. Eur. Respir. J. 2017, 49, 1600965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baz-Redón, N.; Rovira-Amigo, S.; Paramonov, I.; Castillo-Corullón, S.; Roig, M.C.; Antolín, M.; Arumí, E.G.; Torrent-Vernetta, A.; Messa, I.D.M.; Gartner, S.; et al. Implementation of a Gene Panel for Genetic Diagnosis of Primary Ciliary Dyskinesia. Arch. Bronconeumol. 2020, 20, 30073–30079. [Google Scholar] [CrossRef]
- Kempeneers, C.; Seaton, C.; Espinosa, B.G.; Chilvers, M.A. Ciliary Functional Analysis: Beating a Path towards Standardization. Pediatric Pulmonol. 2019, 54, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Kouis, P.; Yiallouros, P.; Middleton, N.; Evans, J.S.; Kyriacou, K.; Papatheodorou, S.I. Prevalence of Primary Ciliary Dyskinesia in Consecutive Referrals of Suspect Cases and the Transmission Electron Microscopy Detection Rate: A Systematic Review and Meta-Analysis. Pediatric Res. 2016, 81, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Fliegauf, M.; Olbrich, H.; Horvath, J.; Wildhaber, J.H.; Zariwala, M.A.; Kennedy, M.; Knowles, M.R.; Omran, H. Mislocalization of DNAH5 and DNAH9 in Respiratory Cells from Patients with Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2005, 171, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Baz-Redón, N.; Rovira-Amigo, S.; Camats-Tarruella, N.; Fernández-Cancio, M.; Garrido-Pontnou, M.; Antolín, M.; Reula, A.; Armengot-Carceller, M.; Carrascosa, A.; Moreno-Galdó, A. Role of Immunofluorescence and Molecular Diagnosis in the Characterization of Primary Ciliary Dyskinesia. Arch. Bronconeumol. 2019, 55, 439–441. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Fenske, L.; Mussaffi, H.; Horvath, J.; Fliegauf, M.; Kuhl, H.; Baktai, G.; Peterffy, E.; Chodhari, R.; et al. DNAI2 Mutations Cause Primary Ciliary Dyskinesia with Defects in the Outer Dynein Arm. Am. J. Hum. Genet. 2008, 83, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallmeier, J.; Shiratori, H.; Dougherty, G.W.; Edelbusch, C.; Hjeij, R.; Loges, N.T.; Menchen, T.; Olbrich, H.; Pennekamp, P.; Raidt, J.; et al. TTC25 Deficiency Results in Defects of the Outer Dynein Arm Docking Machinery and Primary Ciliary Dyskinesia with Left-Right Body Asymmetry Randomization. Am. J. Hum. Genet. 2016, 99, 460–469. [Google Scholar] [CrossRef]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.; et al. CCDC151 Mutations Cause Primary Ciliary Dyskinesia by Disruption of the Outer Dynein Arm Docking Complex Formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Loges, N.T.; Antony, D.; Maver, A.; Deardorff, M.A.; Güleç, E.Y.; Gezdirici, A.; Nöthe-Menchen, T.; Höben, I.M.; Jelten, L.; Frank, D.; et al. Recessive DNAH9 Loss-of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects. Am. J. Hum. Genet. 2018, 103, 995–1008. [Google Scholar] [CrossRef] [Green Version]
- Fassad, M.R.; Shoemark, A.; Legendre, M.; Hirst, R.A.; Koll, F.; Le Borgne, P.; Louis, B.; Daudvohra, F.; Patel, M.P.; Thomas, L.; et al. Mutations in Outer Dynein Arm Heavy Chain DNAH9 Cause Motile Cilia Defects and Situs Inversus. Am. J. Hum. Genet. 2018, 103, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is Required for Axonemal Dynein Assembly and Ciliary Motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef]
- Knowles, M.R.; Ostrowski, L.E.; Loges, N.T.; Hurd, T.; Leigh, M.W.; Huang, L.; Wolf, W.E.; Carson, J.L.; Hazucha, M.J.; Yin, W.; et al. Mutations in SPAG1 Cause Primary Ciliary Dyskinesia Associated with Defective Outer and Inner Dynein Arms. Am. J. Hum. Genet. 2013, 93, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Olcese, C.; UK10K Rare Group; Patel, M.P.; Shoemark, A.; Kiviluoto, S.; Legendre, M.; Williams, H.J.; Vaughan, C.K.; Hayward, J.; Goldenberg, A.; et al. X- Linked Primary Ciliary Dyskinesia Due to Mutations in the Cytoplasmic Axonemal Dynein Assembly Factor PIH1D3. Nat. Commun. 2017, 8, 14279. [Google Scholar] [CrossRef] [Green Version]
- Paff, T.; Loges, N.T.; Aprea, I.; Wu, K.; Bakey, Z.; Haarman, E.G.; Daniels, J.M.; Sistermans, E.A.; Bogunovic, N.; Dougherty, G.W.; et al. Mutations in PIH1D3 Cause X-Linked Primary Ciliary Dyskinesia with Outer and Inner Dynein Arm Defects. Am. J. Hum. Genet. 2017, 100, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Loges, N.T.; Olbrich, H.; Becker-Heck, A.; Häffner, K.; Heer, A.; Reinhard, C.; Schmidts, M.; Kispert, A.; Zariwala, M.A.; Leigh, M.W.; et al. Deletions and Point Mutations of LRRC50 Cause Primary Ciliary Dyskinesia Due to Dynein Arm Defects. Am. J. Hum. Genet. 2009, 85, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Austin-Tse, C.; Halbritter, J.; Zariwala, M.A.; Gilberti, R.M.; Gee, H.Y.; Hellman, N.; Pathak, N.; Liu, Y.; Panizzi, J.R.; Patel-King, R.S.; et al. Zebrafish Ciliopathy Screen Plus Human Mutational Analysis Identifies C21orf59 and CCDC65 Defects as Causing Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2013, 93, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchison, H.; Schmidts, M.; Loges, N.T.; Freshour, J.; Dritsoula, A.; Hirst, R.A.; O’Callaghan, C.; Blau, H.; Al Dabbagh, M.; Olbrich, H.; et al. Mutations in Axonemal Dynein Assembly Factor DNAAF3 Cause Primary Ciliary Dyskinesia. Nat. Genet. 2012, 44, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. ZMYND10 Is Mutated in Primary Ciliary Dyskinesia and Interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diggle, C.P.; Toddie-Moore, D.; Mali, G.; Lage, P.Z.; Ait-Lounis, A.; Schmidts, M.; Shoemark, A.; Munoz, A.G.; Halachev, M.R.; Gautier, P.; et al. HEATR2 Plays a Conserved Role in Assembly of the Ciliary Motile Apparatus. PLoS Genet. 2014, 10, e1004577. [Google Scholar] [CrossRef] [Green Version]
- Horani, A.; Druley, T.E.; Zariwala, M.A.; Patel, A.C.; Levinson, B.T.; Van Arendonk, L.G.; Thornton, K.C.; Giacalone, J.C.; Albee, A.J.; Wilson, K.S.; et al. Whole-Exome Capture and Sequencing Identifies HEATR2 Mutation as a Cause of Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2012, 91, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassad, M.R.; Shoemark, A.; Le Borgne, P.; Koll, F.; Patel, M.; Dixon, M.; Hayward, J.; Richardson, C.; Frost, E.; Jenkins, L.; et al. C11orf70 Mutations Disrupting the Intraflagellar Transport-Dependent Assembly of Multiple Axonemal Dyneins Cause Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2018, 102, 956–972. [Google Scholar] [CrossRef] [Green Version]
- Höben, I.M.; Hjeij, R.; Olbrich, H.; Dougherty, G.W.; Nöthe-Menchen, T.; Aprea, I.; Frank, D.; Pennekamp, P.; Dworniczak, B.; Wallmeier, J.; et al. Mutations in C11orf70 Cause Primary Ciliary Dyskinesia with Randomization of Left/Right Body Asymmetry Due to Defects of Outer and Inner Dynein Arms. Am. J. Hum. Genet. 2018, 102, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Merveille, A.-C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.W.; Beydon, N.; Billen, F.; Clément, A.; et al. CCDC39 is Required for Assembly of Inner Dynein Arms and the Dynein Regulatory Complex and for Normal Ciliary Motility in Humans and Dogs. Nat. Genet. 2011, 43, 72–78. [Google Scholar] [CrossRef]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The Coiled-Coil Domain Containing Protein CCDC40 Is Essential for Motile Cilia Function and Left-Right Axis Formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Antony, D.; Becker-Heck, A.; Zariwala, M.A.; Schmidts, M.; Onoufriadis, A.; Forouhan, M.; Wilson, R.; Taylor-Cox, T.; Dewar, A.; Jackson, C.; et al. Mutations in CCDC39 and CCDC40 are the Major Cause of Primary Ciliary Dyskinesia with Axonemal Disorganization and Absent Inner Dynein Arms. Hum. Mutat. 2013, 34, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Olbrich, H.; Cremers, C.; Loges, N.T.; Werner, C.; Nielsen, K.G.; Marthin, J.K.; Philipsen, M.; Wallmeier, J.; Pennekamp, P.; Menchen, T.; et al. Loss-of-Function GAS8 Mutations Cause Primary Ciliary Dyskinesia and Disrupt the Nexin-Dynein Regulatory Complex. Am. J. Hum. Genet. 2015, 97, 546–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirschell, M.; Olbrich, H.; Werner, C.; Tritschler, D.; Bower, R.; Sale, W.S.; Loges, N.T.; Pennekamp, P.; Lindberg, S.; Stenram, U.; et al. The Nexin-Dynein Regulatory Complex Subunit DRC1 Is Essential for Motile Cilia Function in Algae and Humans. Nat. Genet. 2013, 45, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horani, A.; Brody, S.L.; Ferkol, T.W.; Shoseyov, D.; Wasserman, M.G.; Ta-Shma, A.; Wilson, K.S.; Bayly, P.V.; Amirav, I.; Cohen-Cymberknoh, M.; et al. CCDC65 Mutation Causes Primary Ciliary Dyskinesia with Normal Ultrastructure and Hyperkinetic Cilia. PLoS ONE 2013, 8, e72299. [Google Scholar] [CrossRef] [PubMed]
- Frommer, A.; Hjeij, R.; Loges, N.T.; Edelbusch, C.; Jahnke, C.; Raidt, J.; Werner, C.; Wallmeier, J.; Große-Onnebrink, J.; Olbrich, H.; et al. Immunofluorescence Analysis and Diagnosis of Primary Ciliary Dyskinesia with Radial Spoke Defects. Am. J. Respir. Cell Mol. Biol. 2015, 53, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Kott, E.; Legendre, M.; Copin, B.; Papon, J.-F.; Moal, F.D.-L.; Montantin, G.; Duquesnoy, P.; Piterboth, W.; Amram, D.; Bassinet, L.; et al. Loss-of-Function Mutations in RSPH1 Cause Primary Ciliary Dyskinesia with Central-Complex and Radial-Spoke Defects. Am. J. Hum. Genet. 2013, 93, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Castleman, V.H.; Romio, L.; Chodhari, R.; Hirst, R.A.; De Castro, S.C.; Parker, K.A.; Ybot-Gonzalez, P.; Emes, R.D.; Wilson, S.W.; Wallis, C.; et al. Mutations in Radial Spoke Head Protein Genes RSPH9 and RSPH4A Cause Primary Ciliary Dyskinesia with Central-Microtubular-Pair Abnormalities. Am. J. Hum. Genet. 2009, 84, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, G.W.; Loges, N.T.; Klinkenbusch, J.A.; Olbrich, H.; Pennekamp, P.; Menchen, T.; Raidt, J.; Wallmeier, J.; Werner, C.; Westermann, C.; et al. DNAH11 Localization in the Proximal Region of Respiratory Cilia Defines Distinct Outer Dynein Arm Complexes. Am. J. Respir. Cell Mol. Biol. 2016, 55, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Olbrich, H.; Schmidts, M.; Werner, C.; Onoufriadis, A.; Loges, N.T.; Raidt, J.; Banki, N.F.; Shoemark, A.; Burgoyne, T.; Al Turki, S.; et al. Recessive HYDIN Mutations Cause Primary Ciliary Dyskinesia without Randomization of Left-Right Body Asymmetry. Am. J. Hum. Genet. 2012, 91, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Edelbusch, C.; Cindrić, S.; Dougherty, G.W.; Loges, N.T.; Olbrich, H.; Rivlin, J.; Wallmeier, J.; Pennekamp, P.; Amirav, I.; Omran, H. Mutation of Serine/Threonine Protein Kinase 36 (STK36 ) Causes Primary Ciliary Dyskinesia with a Central Pair Defect. Hum. Mutat. 2017, 38, 964–969. [Google Scholar] [CrossRef]
- Cindrić, S.; Dougherty, G.W.; Olbrich, H.; Hjeij, R.; Loges, N.T.; Amirav, I.; Philipsen, M.C.; Marthin, J.K.; Nielsen, K.G.; Sutharsan, S.; et al. SPEF2-and HYDIN-Mutant Cilia Lack the Central Pair–associated Protein SPEF2, Aiding Primary Ciliary Dyskinesia Diagnostics. Am. J. Respir. Cell Mol. Biol. 2020, 62, 382–396. [Google Scholar] [CrossRef]
- Knowles, M.R.; Leigh, M.W. Primary Ciliary Dyskinesia Diagnosis. Is Color Better Than Black and White? Am. J. Respir. Crit. Care Med. 2017, 196, 9–10. [Google Scholar] [CrossRef]



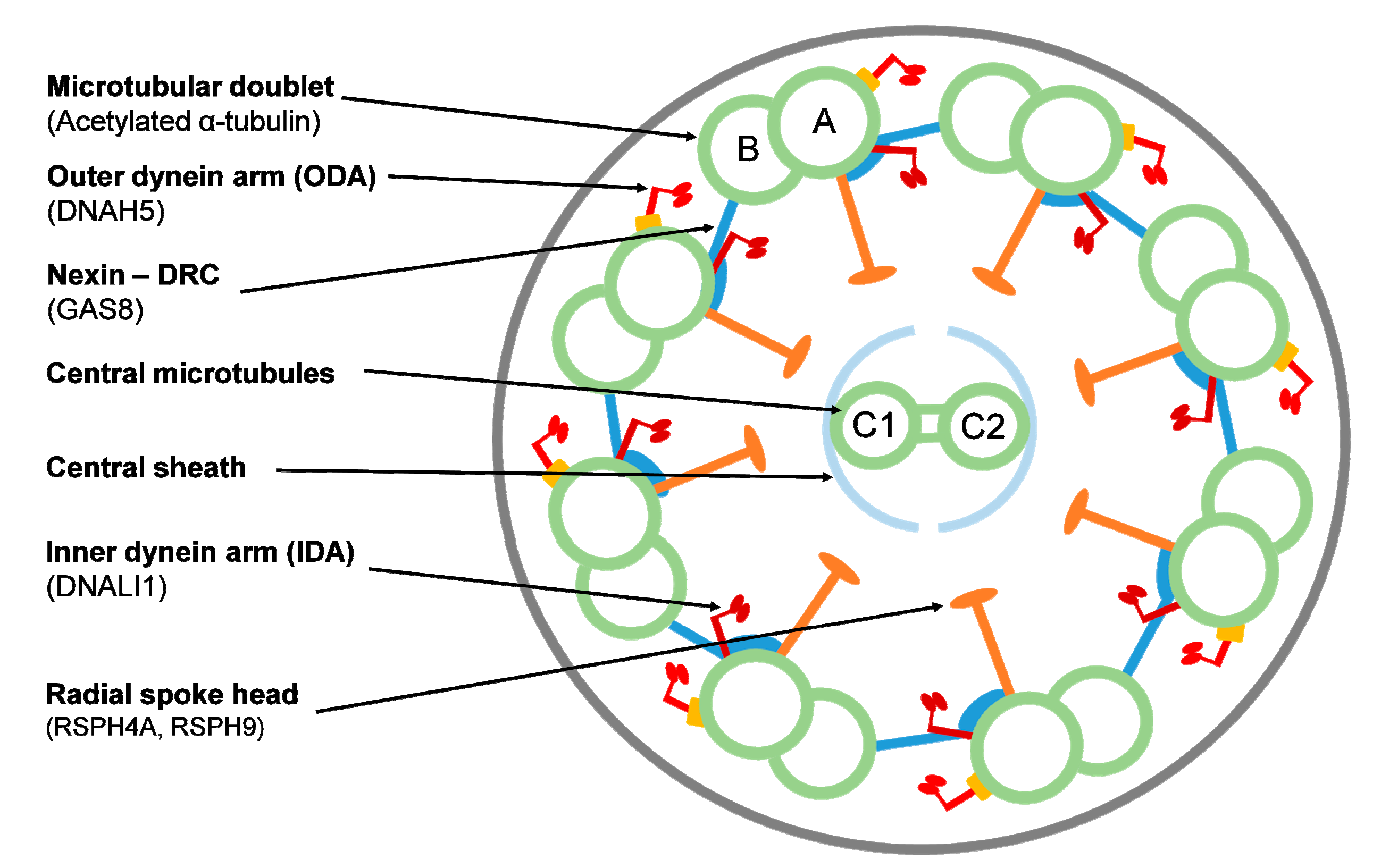{kind=link}
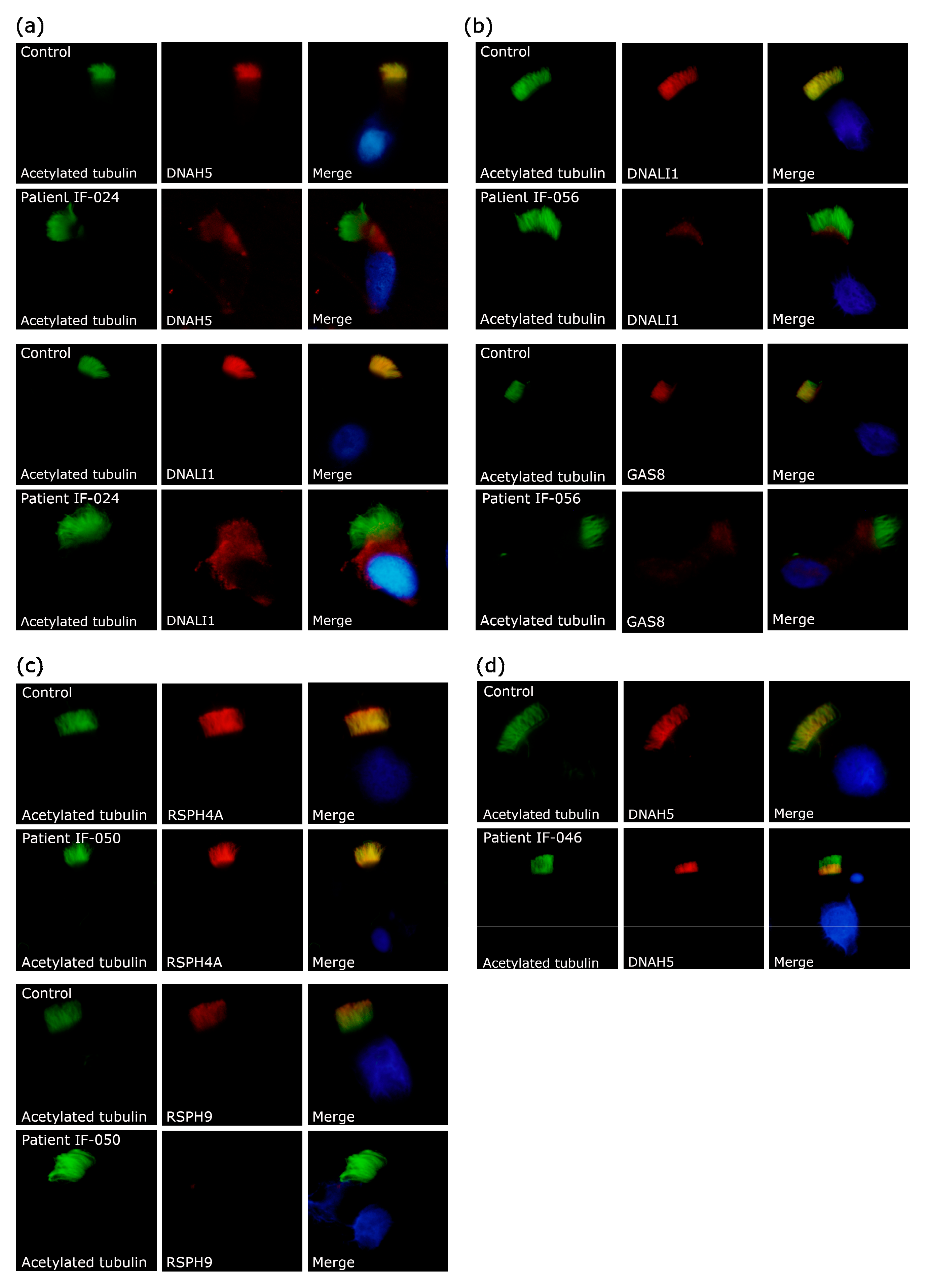{kind=link}
| PCD Diagnostic Evaluation | ||||
|---|---|---|---|---|
| Immunofluorescence Test Outcome (n = 74) | Confirmed (n = 25) | Highly Likely (n = 25) | Highly Unlikely (n = 24) | |
| Evaluable/Closed | 68 (91.9%) | 24 | 24 | 20 |
| Normal results (all markers presents) | 35 (47.3%) | 3 | 12 | 20 |
| Absent/aberrant results | 33 (44.6%) | 21 | 12 | 0 |
| DNAH5 (-) (ODA) | 15 | |||
| Proximal DNAH5 (ODA) | 3 | |||
| DNAH5 (-), DNALI1 (-) (ODA+IDA) | 3 | |||
| DNALI1 (-), GAS8 (-) (IDA+Nexin-DRC) | 7 | |||
| GAS8 (-) (Nexin-DRC) | 1 | |||
| RSPH9 (-) (Radial spoke) | 3 | |||
| RSPH4A (-) (Radial spoke) | 1 | |||
| Inconclusive/insufficient results | 6 (8.1%) | 1 | 1 | 4 |
| IF Affected Markers (Ultrastructural Part) | # | HSVM | Genetics (#) | PCD Symptoms | |||||
|---|---|---|---|---|---|---|---|---|---|
| Neonatal Distress | Upper Respiratory Tract | Lower Respiratory Tract | Bronchiectasis | Chronic Otitis or Hearing loss | Situs Abnormality | ||||
| DNAH5 (ODA) | 15 | Completely immotile cilia or residual motility | CCDC151 (1), DNAH5 (5), DNAI2 (4), TTC25 (1), NA (4) | +/− | + | + | +/− | +/− | +/− |
| Proximal DNAH5 (ODA) | 3 | Subtle defects (stiff and disorganized ciliary beat) | DNAH9 (1), Neg. (2) | - | + | +/− | +/− | +/− | +/− |
| DNAH5+DNALI1 (ODA+IDA) | 3 | Completely immotile cilia | Neg. (2), NA (1) | + | + | + | + | + | +/− |
| DNALI1+GAS8 (IDA+Nexin-DRC) | 7 | Mainly stiff cilia and immotile cilia | CCDC39 (3), CCDC40 (3), NA (1) | +/− | + | +/− | +/− | +/− | +/− |
| GAS8 (Nexin-DRC) | 1 | Hyperkinetic stiff cilia | NA (1) | + | + | + | + | + | − |
| RSPH4A or RSPH9 (Radial spoke) | 4 | Stiff and circular motion | RSPH1 (1), RSPH4A (1), RSPH9 (1), NA (1) | +/− | + | +/− | +/− | +/− | − |
| All markers present (normal result) | 3 | Hyperkinetic stiff cilia | DNAH11 (3) | +/− | + | +/− | +/− | +/− | +/− |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baz-Redón, N.; Rovira-Amigo, S.; Fernández-Cancio, M.; Castillo-Corullón, S.; Cols, M.; Caballero-Rabasco, M.A.; Asensio, Ó.; Martín de Vicente, C.; Martínez-Colls, M.d.M.; Torrent-Vernetta, A.; et al. Immunofluorescence Analysis as a Diagnostic Tool in a Spanish Cohort of Patients with Suspected Primary Ciliary Dyskinesia. J. Clin. Med. 2020, 9, 3603. https://doi.org/10.3390/jcm9113603
Baz-Redón N, Rovira-Amigo S, Fernández-Cancio M, Castillo-Corullón S, Cols M, Caballero-Rabasco MA, Asensio Ó, Martín de Vicente C, Martínez-Colls MdM, Torrent-Vernetta A, et al. Immunofluorescence Analysis as a Diagnostic Tool in a Spanish Cohort of Patients with Suspected Primary Ciliary Dyskinesia. Journal of Clinical Medicine. 2020; 9(11):3603. https://doi.org/10.3390/jcm9113603
Chicago/Turabian StyleBaz-Redón, Noelia, Sandra Rovira-Amigo, Mónica Fernández-Cancio, Silvia Castillo-Corullón, Maria Cols, M. Araceli Caballero-Rabasco, Óscar Asensio, Carlos Martín de Vicente, Maria del Mar Martínez-Colls, Alba Torrent-Vernetta, and et al. 2020. "Immunofluorescence Analysis as a Diagnostic Tool in a Spanish Cohort of Patients with Suspected Primary Ciliary Dyskinesia" Journal of Clinical Medicine 9, no. 11: 3603. https://doi.org/10.3390/jcm9113603